- 印刷用
- 2013年6月25日
エーザイ株式会社(本社:東京、社長:内藤晴夫)は、抗てんかん剤であるAMPA受容体拮抗剤「Fycompa®」(一般名:ペランパネル)について、ドイツで販売承認後に実施される追加有用性の評価において、ドイツ連邦合同委員会(G-BA)が本剤の革新的新薬としての価値を適切に評価していないとして、このたび、本剤のドイツ国内での販売を一時中断することを決定しました。当社は本剤による治療を必要とされている患者様を第一義に考え、販売の一時中断後は、本剤へのアクセスを可能とするPatient Access Program(患者支援プログラム)を提供してまいります。
AMNOG(医薬品市場再編法)に基づく本剤の追加有用性の評価において、G-BAは、2013年3月、G-BAが指定した2つの医薬品と比較したデータが不十分として本剤の追加有用性が証明できていないと結論づけました。しかし、この比較方法について、ドイツてんかん学会ならびにドイツ神経学会は、単剤療法では効果不十分な難治性てんかんに対する併用治療薬については、既存治療薬に抵抗性のある患者様に対する上乗せ(add-on)効果の有無を追加有用性として評価すべきであり、G-BAが求めている既存治療薬との直接的比較による評価は臨床実態を反映しておらず不適切だと指摘しています。また、難治性てんかんの患者様にとって、てんかん発作抑制や副作用を改善する新薬は希望であり、臨床現場では新たな作用機序を持つ本剤の革新性が強く望まれていると訴えています。
本追加有用性評価の制度については、その一部見直しに向けてドイツ医薬品法改正が検討されています。当社は、本剤の販売一時中断を余儀なくされている事態を一日も早く解消するためにも、適切な評価方法に基づいた本剤の再評価が早急に行われ、本剤の有用性ならびに革新性が認められることを強く望んでいます。
本剤は、2012年7月、12 歳以上のてんかん患者様の部分発作(二次性全般化発作を含む)に対する併用療法として欧州委員会から承認され、ドイツでは同年9月より販売を開始し、これまでに3,000人以上の患者様に使用されています。現在、欧州7カ国で販売されています。米国では、同年10 月に米国食品医薬品局(FDA)より承認を取得し、米国麻薬取締局(DEA)による規制物質法に基づくスケジュール審査完了後に発売予定です。
当社は、革新的新薬を必要とされているドイツの部分てんかん患者様のもとに、本剤を確実にお届けできるように、引き続き、関係当局に本剤の価値に対する正しい理解と評価を求めてまいります。
以上
[参考資料として、てんかん治療、用語解説、Fycompa®、臨床第Ⅲ相試験について、添付しています]
<参考資料>
1. てんかんの治療について
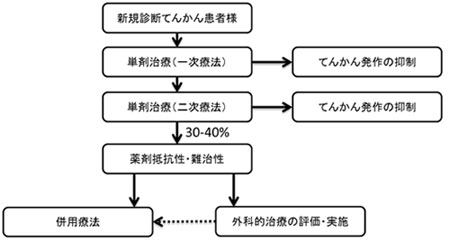
てんかん患者様の治療ゴールはてんかん発作の抑制です。てんかんと診断されると抗てんかん薬の単剤療法で治療を行いますが、発作抑制効果が十分でない場合には、他の単剤療法に切り替えます。しかし、約3~4割の患者様は、2種類の単剤療法を試みても発作を抑制できないことが報告されており、その場合には併用療法に移行します。「Fycompa®」のようにてんかんの併用治療薬として承認を取得している薬剤は、単剤治療薬へのadd-on(上乗せ)として使用されます。症状によっては、更に複数の併用治療薬がadd-onされる場合もあります。
2. 用語解説
- 1)ドイツ連邦合同委員会(G-BA)
医師、歯科医師、病院および疾病金庫からなる独立したドイツにおける医療制度の最高意思決定機関であり、医薬品をはじめとする医療サービスに関する保険償還などについての決定権を有しています。
- 2)AMNOG (Arzneimittelmarkt-Neuordnungsgesetz: 医療品市場再編法)について
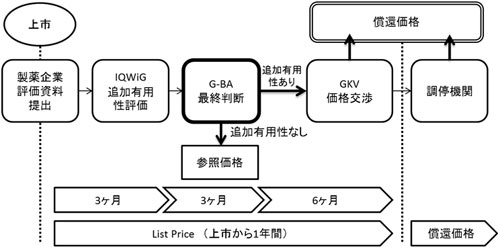
医薬品市場再編法(AMNOG)は、2011年1月に施行されました。これにより、ドイツで発売された全ての新薬は、G-BAによる追加有用性評価を受けることが義務付けられ、この評価に基づく価格交渉を経て、発売後1年以内に償還価格が決定されることになりました。
新製品上市に際して、製薬企業はG-BAが定めた比較治療薬に対して追加的な有用性を示した提出書類(benefit dossier)を提出する必要があります。提出された資料は、G-BAからの委託を受けたIQWiGにより比較治療薬に対する追加有用性があるか評価され、公表されます。製薬企業に対して、本評価に対するヒアリングによるコメントの機会が与えられた後、G-BAが当該薬剤に対する有用性評価に対する最終判断を行います。
G‐BAによる追加有用性が認められた場合には、公的医療保険協会(GKV-SV)との価格交渉段階に進み、G-BAによって設定された有用性レベルに基づき償還価格が決定されます。一方、追加有用性が認められないと判断された薬剤には、原則参照価格が適応され、評価で用いられた比較治療薬と同レベルの価格に設定されます。
3. 「Fycompa®」(ペランパネル)について
「Fycompa®」(ペランパネル)は、当社が創製した新規化合物であり、AMPA受容体に対して非競合的な拮抗剤です。本剤は、シナプス後AMPA受容体のグルタミン酸による活性化を阻害し、神経の過興奮を抑制することで、抗てんかん作用を発揮すると考えられています。本剤は、部分てんかんを対象とした臨床第Ⅱ相および第Ⅲ相試験において、発作抑制効果が示されています。本剤は、1日1回の経口投与の部分てんかん治療剤として、欧州並びに米国で承認を取得、および日本・中国・アジアでは臨床第Ⅲ相試験の段階にあります。さらなる適応の拡大をめざし、全般てんかんについて国際共同治験として臨床第Ⅲ相試験を、部分てんかんの小児患者様を対象に欧米で臨床第Ⅱ相試験を実施しています。
4. ペランパネルの臨床第Ⅲ相試験について
ペランパネルの部分てんかんの申請用試験は3つの臨床第Ⅲ相試験(304、305、および306試験)から成り、計1,480人の青年期を含む12歳以上の部分てんかん患者様が本試験に参加されました。306試験は、最小有効量を把握することを主目的として、「プラセボ、2mg、4mg、8mg」の4群で実施されました。また、304試験と305試験は、投与量範囲の決定を主目的とし、「プラセボ、8mg、12mg」の3群で実施されました。
いずれの試験も、グローバル、無作為化、プラセボ対照、二重盲検、並行群間比較、用量漸増試験として実施され、「部分発作回数変化率(percentage change in seizure frequency)」、「50%レスポンダーレート(50% responder rate、部分発作回数が観察期間と比べて50%以上改善した症例の割合)」、「複雑部分発作および二次性全般化発作減少率(percentage reduction of complex partial plus secondarily generalized seizures)」、および「用量反応性(evaluation for dose response)」を評価項目としていました。また、EMAの主要評価項目には「50%レスポンダーレート」が、米国FDAの主要評価項目には「部分発作回数変化率(中央値)」が設定されました。各試験の全症例を対象とした結果は下記の通りです。
- 1)306試験
- 「50%レスポンダーレート」:プラセボ投与群の17.9%に対し、ペランパネルの2mg投与群で20.6%
(p=0.4863)、4mg投与群で28.5%(p=0.0132)、8 mg投与群で34.9%(p=0.0003) - 「発作回数変化率(中央値)」:プラセボ投与群の-10.7%に対し、ペランパネルの2mg投与群で-13.6%
(p=0.4197)、4mg投与群で-23.3%(p=0.0026)、8 mg投与群で-30.8%(p<0.0001) - 「主な有害事象」:めまい、頭痛、眠気
- 「50%レスポンダーレート」:プラセボ投与群の17.9%に対し、ペランパネルの2mg投与群で20.6%
- 2)305試験
- 「50%レスポンダーレート」:プラセボ投与群の14.7%に対し、ペランパネルの8mg投与群で33.3%
(p=0.0018)、12mg投与群で33.9%(p=0.0006) - 「発作回数変化率(中央値)」:プラセボ投与群の-9.7%に対して、ペランパネルの8mg投与群で-30.5%
(p=0.0008)、12mg投与群で-17.6%(p=0.0105) - 「主な有害事象」:めまい、けん怠感、頭痛、眠気
- 「50%レスポンダーレート」:プラセボ投与群の14.7%に対し、ペランパネルの8mg投与群で33.3%
- 3)304試験
- 「50%レスポンダーレート」:プラセボ投与群の26.4%に対し、ペランパネルの8mg投与群で37.6%
(p=0.0760)、12mg投与群で36.1%(p=0.0914) - 「発作回数変化率(中央値)」:プラセボ投与群の-21.0%に対して、ペランパネルの8mg投与群で-26.3%
(p=0.0261)、12mg投与群で-34.5%(p=0.0158) - 「主な有害事象」:めまい、眠気、神経過敏、頭痛、転倒、運動失調
- 「50%レスポンダーレート」:プラセボ投与群の26.4%に対し、ペランパネルの8mg投与群で37.6%