- 印刷用
- 2022年11月30日
エーザイ株式会社
バイオジェン・インク
エーザイ株式会社(本社:東京都、代表執行役CEO:内藤晴夫、以下 エーザイ)とバイオジェン・インク (Nasdaq: BIIB、本社:米国マサチューセッツ州ケンブリッジ、CEO:Christopher A. Viehbacher、以下 バイオジェン)は、本日、抗アミロイドβ(Aβ)プロトフィブリル抗体レカネマブ(開発品コード:BAN2401)について、脳内アミロイド病理が確認されたアルツハイマー病(AD)による軽度認知障害(Mild Cognitive Impairment:MCI)および軽度AD(これらを総称して早期ADと定義)を対象とした大規模なグローバル臨床第Ⅲ相Clarity AD検証試験の結果を米国カリフォルニア州サンフランシスコおよびバーチャルで開催されている第15回アルツハイマー病臨床試験会議(CTAD:Clinical Trials on Alzheimer’s Disease)において発表することをお知らせします。
CTADサイエンティフィック・セッションにおけるレカネマブのデータ発表の要旨
●Clarity AD試験デザイン
本試験は、北米、欧州、アジアの235施設で早期AD当事者様1,795人(レカネマブ投与群:898人、プラセボ投与群:897人)を対象とした、プラセボ対照、二重盲検、並行群間比較、無作為化グローバル臨床第Ⅲ相検証試験です。被験者は、レカネマブ投与群(10 mg/kg bi-weekly静脈投与)またはプラセボ投与群に1:1で割り付けられ、疾患ステージ(ADによるMCIまたは軽度AD)、AD症状改善薬併用の有無(例:アセチルコリンエステラーゼ阻害薬、メマンチンまたはその両方)、ApoE4ステータス、および地域によって層別割付けされました。被験者登録基準においては、多様な合併症あるいは併用治療(高血圧症、糖尿病、心疾患、肥満、腎臓病、抗凝固剤併用など)を許容しています。本試験では、民族的・人種的多様性を考慮した被験者登録を推進した結果、米国では登録被験者の4.5%が黒人、22.5%がヒスパニック系となりました。
主要評価項目は、全般臨床症状の評価指標であるCDR-SB1(Clinical Dementia Rating Sum of Boxes)の18カ月時点におけるベースラインからの変化とし、重要な副次評価項目として、アミロイドポジトロン断層法(PET)測定(センチロイド法)による脳内アミロイド蓄積、ADAS-Cog142(Alzheimer's Disease Assessment Scale-Cognitive subscale 14)、ADCOMS3(Alzheimer’s Disease Composite Score)およびADCS MCI-ADL4(Alzheimer's Disease Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment)の投与18カ月時点でのベースラインからの変化が設定されました。また、タウPETで測定する脳内タウ病理(n=257)、脳脊髄液(CSF)バイオマーカー(n=281)を評価しました。
●Clarity AD試験の有効性評価結果
主要評価項目である投与18カ月時点のCDR-SBスコアのベースラインからの平均変化量について、レカネマブ投与群、プラセボ投与群はそれぞれ1.21、1.66であり、その変化量の差は-0.45([95%信頼区間(CI): -0.67, -0.23];P = 0.00005)となり統計学的に高度に有意な結果が認められ、レカネマブ投与群はプラセボ投与群と比較して27%の全般臨床症状の悪化抑制を示しました。CDR-SBスコアの平均変化量の差は、投与6カ月時点(変化量の差:-0.17 [95%CI:-0.29, -0.05];P<0.01)から3カ月毎のすべての評価時点においてプラセボ投与群と比較して統計学的に高度に有意な結果を示し、その絶対値は経時的に拡大を示しました(全評価ポイントでp<0.01)(図1)。
また、すべての重要な副次評価項目において、レカネマブ投与群はプラセボ投与群と比較して統計学的に高度に有意な結果(P<0.001)が認められました。アミロイドPET評価では、レカネマブ投与後3カ月からすべての評価時点で、統計学的に有意な脳内アミロイド蓄積の減少がみられ、投与18カ月時点のレカネマブ投与群における脳内アミロイドの平均変化量(センチロイド)は-55.5、プラセボ投与群で3.6(平均差:-59.1 [95% CI:-62.6, -55.6]; P<0.00001 )でした。投与18カ月時点でのADAS-Cog14評価では26%の認知機能低下の抑制(平均差: -1.44[95%CI:-2.27, -0.61];P=0.00065 )、ADCOMSでは24%の疾患進行の抑制(平均差:-0.050[95%CI:-0.074, -0.027];P=0.00002)、ADCS MCI-ADLでは37%の日常生活動作低下の抑制(平均差:2.016 [95%CI:1.208, 2.823];P<0.00001)を示しました。さらに、主要な層別解析では、疾患ステージ(ADによるMCIまたは軽度AD)、ApoE4ステータス(非保持者、保持者)、AD症状改善薬併用の有無、および地域(北米、アジア、欧州)のいずれのサブグループにおいても、レカネマブ投与18カ月時点のCDR-SB、ADAS-Cog14、ADCS MCI-ADLについて一貫した悪化抑制が認めらました。
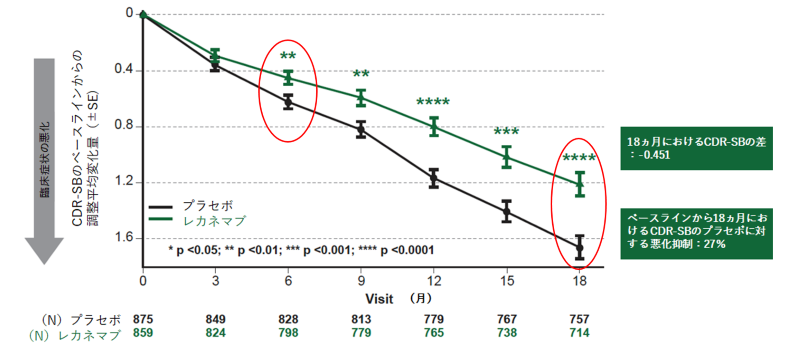
図1:主要評価項目CDR-SBのレカネマブ投与による変化(18カ月)
●Clarity AD試験の安全性評価結果
レカネマブ投与群で最も多かった有害事象(10%以上)は、静脈注入に伴う反応(レカネマブ:26.4%、プラセボ:7.4%)、ARIA-H(ARIAによる脳微小出血、大出血、脳表ヘモジデリン沈着)(レカネマブ:17.3%、プラセボ:9.0%)、ARIA-E(浮腫/浸出)(レカネマブ:12.6%、プラセボ:1.7%)、頭痛(レカネマブ:11.1%、プラセボ:8.1%)および転倒(レカネマブ:10.4%、プラセボ:9.6%)でした。静脈注入に伴う反応は大部分が軽度から中等度(グレード1-2:96%)であり、その多くは初回投与時に発現しました(75%)。本試験の18カ月の二重盲検試験期間中における死亡例はレカネマブ投与群で0.7%、プラセボ投与群で0.8%でしたが、レカネマブやアミロイド関連画像異常(ARIA)発現に関連する死亡例はありませんでした。重篤な有害事象の発現率は、レカネマブ投与群で14.0%、プラセボ投与群で11.3%でした。治験薬投与後に発現した有害事象(TEAEs)は、レカネマブ投与群で88.9%、プラセボ投与群で81.9%でした。投薬中止に至ったTEAEsは、レカネマブ投与群で6.9%、プラセボ投与群で2.9%でした。
レカネマブのARIA発現プロファイルは、臨床第Ⅱ相試験(201試験)結果を踏まえ、総じて想定内でした。ARIA-Eは、画像診断では大部分が軽度から中等度(ARIA -E発現者の91%)であり、無症状(同78%)でした。その多くは治療開始後3カ月以内に発現(同71%)し、発見後4カ月以内に消失(同81%)しました。症候性のARIA-Eの発現率は、レカネマブ投与群で2.8%であり、最も一般的な症状として頭痛、視覚障害、錯乱が報告されました。症候性ARIA-Hの発現率は、レカネマブ投与群で0.7%、プラセボ投与群で0.2%でした。ARIA-Hのみ(ARIA-Eを発現していない被験者でのARIA-H)は、レカネマブ投与群(8.9%)、プラセボ投与群(7.8%)であり、差はありませんでした。ARIA-EおよびARIA-Hは、ApoE4非保持者でApoE4保持者より発現頻度が低く、またApoE4ホモ接合体保持者ではApoE4ヘテロ接合体保持者よりも高い頻度で観察されました。なお、コア試験とその後の非盲検長期投与試験において、大出血が併発した死亡率は、プラセボ投与群(1/897例)、レカネマブ投与群(2/1608例)ともに0.1%でした。レカネマブ投与群の2例は非盲検長期投与試験において発生し、いずれも重大な合併症および大出血や死亡の一因となる抗凝固薬使用などのリスク因子を有していたため、エーザイはこれらをレカネマブに起因する死亡ではないと評価しました。
●Clarity AD試験の画像、血漿、CSFバイオマーカー評価結果
レカネマブ投与によるアミロイド、タウ、神経変性に関する画像、血漿、CSFを用いたバイオマーカー評価を行いました。アミロイド関連のバイオマーカーでは、レカネマブ投与により、CSFおよび血漿中Aβ 42/40比において早期より持続したアミロイド除去効果が示されました。アミロイドPET評価では、レカネマブ投与18カ月時点の脳内アミロイドレベルの平均値が22.99センチロイドとなり、アミロイド陽性の閾値30センチロイドを下回りました。タウ関連のバイオマーカーでは、アミロイドの除去が進むと、AD病理のパスウェイのアミロイドの下流にあたるCSFおよび血漿中リン酸化タウ(p-tau181)においても改善することが示されました。タウPETによる解析では、レカネマブ投与によりプラセボ群と比較して、側頭葉へのタウの蓄積が遅くなることも確認されました。CSF中総タウ蛋白(t-tau)はレカネマブ投与で改善しました。神経変性に関するバイオマーカーについては、CSFおよび血漿中のニューロフィラメント軽鎖についてはレカネマブ投与とプラセボ投与による顕著な差は確認されませんでしたが、アストロサイトの活性化マーカーである血漿中GFAP(glial fibrillary acidic protein)とシナプス機能不全のマーカーであるCSF中ニューログラニンはレカネマブ投与により正常な方向へと改善されました。
●Clarity AD試験結果の臨床的意義
ADは進行性の神経疾患であり、高齢化の進展とともに当事者様、ご家族、ヘルスケアシステムに大きな影響を与える社会的課題となっており、疾患病理に作用する新たな治療薬が求められています。早期ADを対象とした治療のゴールは認知機能や日常生活機能、精神症状への持続的な効果や進行抑制による自立の維持、QOLの改善・維持です。
レカネマブは、Clarity AD検証試験において複数の認知機能・全般症状の指標で一貫した悪化抑制を示し、サブグループ(人種、民族、合併症)における臨床効果も一貫性を示しました。また、レカネマブ投与によりCDRの重症度評価でより後期の疾患ステージへの進行リスクを31%低減します(Hazard Ratio: 0.69)。CDR-SBの観察データと30カ月までの外挿に基づく傾斜分析によると、投与後18カ月時点のプラセボ群と同じレベルに達するにはレカネマブ投与では25.5カ月かかり、7.5カ月の進行抑制が示されました。201試験結果に基づくモデルシミュレーションによると、レカネマブはより軽度なADステージにある期間を2.5年~3.1年延長する可能性が示唆され、早期ステージの状態をより長く維持することが期待されます。さらに、AD当事者様の健康関連QOLの維持や介護者様の負担軽減(23-56%のスコア悪化抑制)も示されました。これらの結果から、レカネマブによる治療が早期AD当事者様とそのご家族、医療従事者、社会にとって、意義のあるベネフィットをもたらすことが期待されます。
これらのレカネマブに関するサイエンティフィック・セッションにおける発表は、エーザイのコーポレートウェブサイトの投資家セクションにてライブ配信されます(英語のみ)。内容は改めてオンデマンドでも配信予定です。
レカネマブについて、エーザイは、開発および薬事申請をグローバルに主導し、エーザイの最終意思決定権のもとで、エーザイとバイオジェンが共同商業化・共同販促を行います。
1 CDR-SBは、認知症の幅広いステージの重症度を評価するスケールであり、記憶、見当識、判断力と問題解決、地域社会の活動、家庭および趣味、身の回りの世話の6項目について、当事者様の診察やご家族および介護者様からの情報で評価します。6項目のスコアの合計点がCDR-SBのスコアとなり、早期ステージのADを対象とした治療薬の適切な有効性評価項目としても使用されます。
2 ADAS-Cogは、ADを対象とした臨床治験でグローバルに最も広く用いられている検査方法です。ADAS-Cog14は単語再生、命令、構成行為、物品と手指の呼称 、観念行為 、見当識、単語再認、検査教示の記憶、話し言葉の理解、換語、話し言葉の能力、単語の遅延再生 、数字の消去、迷路という14項目を評価するもので、MCIを含む早期ADを対象とした試験で用いられています。
3 ADCOMSは早期ADの変化を感度よく検出することを目的とし、ADAS-Cog、MMSE(Mini-Mental State Examination)、CDRの3つの臨床評価尺度を組み合わせたエーザイが開発した評価指標です。
4 ADCS MCI-ADL はMCI当事者様の日常生活動作を評価するスケールであり、最近の日常生活動作における実際の様子を被験者のパートナーへの24項目の質問から評価します。
以上
本件に関する報道関係お問い合わせ先
-
エーザイ株式会社
PR部
TEL:03-3817-5120
-
バイオジェン・インク
パブリック アフェアーズ
public.affairs@biogen.com
- 1. Clarity ADの概要
※左右にスクロールできます
| 試験名称 | 早期アルツハイマー病患者を対象に、レカネマブの安全性及び有効性を検証することを目的とした試験(Clarity AD) |
| 対象 | 脳内アミロイド病理が確認されたアルツハイマー病(AD)による軽度認知障害および軽度AD(総称して早期AD)を対象(グローバルで1,795人、および中国で進行中の 111 人) |
| 投与法 |
レカネマブを10mg/kg bi-weekly投与 |
| 治療期間 | 18カ月 |
| 実施地域 | 日本、米国、欧州、中国、韓国、カナダ、豪州、シンガポール |
| 主要評価項目 | ベースラインから投与18カ月時点でのCDR-SB(Clinical Dementia Rating Sum of Boxes)の変化 |
| 主な副次評価項目 | ベースラインから投与18カ月時点での、アミロイドPET測定による脳内アミロイド蓄積、ADAS-Cog14(Alzheimer's Disease Assessment Scale-Cognitive subscale 14)、ADCOMS(Alzheimer’s Disease Composite Score)およびADCS MCI-ADL(Alzheimer's Disease Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment) |
| 解析対象 | 有効性解析は、少なくとも1回の投与を受け、ベースライン評価と少なくとも1回の投与後主要有効性測定を行った無作為化被験者集団と定義した修正intention-to-treat集団1,734人(レカネマブ投与群:859人、 プラセボ投与群:875人)で実施されました。安全性の解析は無作為化された1,795人全員(レカネマブ投与群:898人、プラセボ投与群:897人)で行いました。 |
2. レカネマブについて
レカネマブは、BioArctic AB(本社:スウェーデン、以下 バイオアークティック)とエーザイの共同研究から得られた、可溶性のアミロイドβ(Aβ)凝集体(プロトフィブリル)に対するヒト化モノクローナル抗体です。レカネマブは、ADを惹起させる因子の一つと考えられている、神経毒性を有するAβプロトフィブリルに選択的に結合して無毒化し、脳内からこれを除去することでADの病態進行を抑制する疾患修飾作用が示唆されています。現在、レカネマブは抗Aβ抗体で唯一漸増投与が不要な早期AD治療薬として開発中です。早期ADを対象とした大規模臨床第Ⅱ相試験(201試験)において、事前に規定したレカネマブ10mg/kg bi-weekly 18カ月静脈投与における解析の結果、脳内Aβ蓄積量の減少(p<0.0001)とADCOMSによる臨床症状の悪化抑制(p<0.05)を示しました。なお、12カ月投与時における主要評価項目*は達成しませんでした。201試験(コア期間)の後、投与を休止していた9〜59カ月の無投与期間(平均24カ月、参加者180人)を経て、レカネマブ10mg/kg bi-weekly投与の安全性と有効性を評価するOpen-Label Extension試験が進行中です。
2020年7月から、臨床症状は正常で、ADのより早期ステージにあたる脳内Aβ蓄積が境界域レベルおよび陽性レベルのプレクリニカルADを対象とした臨床第Ⅲ相試験(AHEAD 3-45試験)を米国のADおよび関連する認知症の学術的臨床試験のための基盤を提供するAlzheimer's Clinical Trials Consortium(ACTC)とのパブリック・プライベート・パートナーシップ(PPP)で行っています。ACTCは、National Institutes of Health、National Institute on Agingによる資金提供を受けています。
また、2022年1月から、セントルイス・ワシントン大学医学部(米国ミズーリ州セントルイス)が主導する優性遺伝アルツハイマーネットワーク試験ユニット(Dominantly Inherited Alzheimer Network Trials Unit、以下 DIAN-TU)が実施する優性遺伝アルツハイマー病(DIAD)に対する臨床試験(Tau NexGen試験)が進行中です。本試験において、レカネマブは抗Aβ療法による基礎療法として選定されました。
さらに、レカネマブの皮下注射製剤の臨床第Ⅰ相試験が進行中です。
2022年7月、米国において迅速承認制度に基づくレカネマブの生物製剤ライセンス申請(Biologics License Application: BLA)が米国食品医薬品局(FDA)に受理され、現在審査中です。本迅速承認申請は優先審査(Priority Review)の指定を受け、PDUFA(Prescription Drug User Fee Act)アクション・デート(審査終了目標日)が2023年1月6日に定められました。FDAは、Clarity AD試験結果をレカネマブの臨床的有用性の検証試験として評価することに合意しています。迅速承認制度では本検証試験以外の全てのデータが審査され、フル承認に向けた申請においては主に検証試験が審査対象となります。また、日本においても、2022年3月より、医薬品事前評価相談制度を活用し、本検証試験以外の申請データを医薬品医療機器総合機構(PMDA)に提出しています。エーザイは、Clarity AD試験の結果に基づいて、2022年度中の米国におけるフル承認申請、ならびに日本、欧州における販売承認申請をめざし、各国当局と協議を行っています。
* 投与12カ月時点においてADCOMSによる臨床症状の抑制がプラセボ投与群に対し25%低下する確率が80%以上とする
- 3. エーザイとバイオジェンによるAD領域の提携について
エーザイとバイオジェンは、AD治療剤の共同開発・共同販売に関する提携を2014年から行っています。レカネマブについて、エーザイは、開発および薬事申請をグローバルに主導し、エーザイの最終意思決定権のもとで、エーザイとバイオジェンが共同商業化・共同販促を行います。
- 4. エーザイとバイオアークティックによるAD領域の提携について
2005年以来、エーザイとバイオアークティックはAD治療薬の開発と商業化に関して長期的な協力関係を築いてきました。エーザイは、レカネマブについて、2007年12月にバイオアークティックとのライセンス契約により、全世界におけるADを対象とした研究・開発・製造・販売に関する権利を取得しています。2015年5月にレカネマブのバックアップ抗体の開発・商業化契約を締結しました。
- 5. エーザイ株式会社について
エーザイ株式会社は、患者様と生活者の皆様の喜怒哀楽を第一義に考え、そのベネフィット向上に貢献する「ヒューマン・ヘルスケア(hhc)」を企業理念とし、この理念のもと、人々の「健康憂慮の解消」や「医療較差の是正」という社会善を効率的に実現することをめざしています。グローバルな研究開発・生産・販売拠点ネットワークを持ち、戦略的重要領域と位置づける「神経領域」「がん領域」を中心とするアンメット・メディカル・ニーズの高い疾患領域において、革新的な新薬の創出と提供に取り組んでいます。
また、当社は、国連の持続可能な開発目標(SDGs)のターゲット(3.3)である「顧みられない熱帯病(NTDs)」の制圧に向けた活動に世界のパートナーと連携して積極的に取り組んでいます。
エーザイ株式会社の詳細情報は、https://www.eisai.co.jpをご覧ください。Twitterアカウント@Eisai_SDGs でも情報公開しています。
- 6. バイオジェン・インクについて
神経科学領域のパイオニアであるバイオジェンは、最先端の医学と科学を通じて、重篤な神経学的疾患、神経変性疾患の革新的な治療法の発見および開発を行い、その成果を世界中の患者さんに提供しています。1978年にチャールズ・ワイスマン、ハインツ・シェイラー、ケネス・マレー、ノーベル賞受賞者であるウォルター・ギルバートとフィリップ・シャープにより設立されたバイオジェンは、世界で歴史のあるバイオテクノロジー企業のひとつです。バイオジェンは多発性硬化症の領域をリードする製品ポートフォリオを持ち、脊髄性筋萎縮症の最初の治療薬を製品化し、アルツハイマー病の病理に作用する最初で唯一の治療薬を提供しています。また、生物製剤の高い技術力を活かしてバイオシミラーの製品化を行い、業界内で最も多様な神経科学領域のパイプラインに注力し、進展させており、アンメットニーズが高い疾患領域の患者さんの治療水準に変化をもたらしています。
バイオジェンに関する情報については、https://www.biogen.com/ およびSNS媒体Twitter, LinkedIn, Facebook, YouTubeをご覧ください。
Biogen Safe Harbor
This news release contains forward-looking statements, including statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, about the potential clinical effects of lecanemab; the potential benefits, safety and efficacy of lecanemab; potential regulatory discussions, submissions and approvals and the timing thereof; the expected data readout for the Clarity AD study; the treatment of Alzheimer’s disease; the anticipated benefits and potential of Biogen’s collaboration arrangements with Eisai; the potential of Biogen’s commercial business and pipeline programs, including lecanemab; and risks and uncertainties associated with drug development and commercialization. These statements may be identified by words such as “aim,” “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “possible,” “potential,” “will,” “would” and other words and terms of similar meaning. Drug development and commercialization involve a high degree of risk, and only a small number of research and development programs result in commercialization of a product. Results in early-stage clinical studies may not be indicative of full results or results from later stage or larger scale clinical studies and do not ensure regulatory approval. You should not place undue reliance on these statements or the scientific data presented.
These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including without limitation unexpected concerns that may arise from additional data, analysis or results obtained during clinical studies, including the Clarity AD clinical trial and AHEAD 3-45 study; the occurrence of adverse safety events; risks of unexpected costs or delays; the risk of other unexpected hurdles; regulatory submissions may take longer or be more difficult to complete than expected; regulatory authorities may require additional information or further studies, or may fail or refuse to approve or may delay approval of Biogen’s drug candidates, including lecanemab; actual timing and content of submissions to and decisions made by the regulatory authorities regarding lecanemab; uncertainty of success in the development and potential commercialization of lecanemab; failure to protect and enforce Biogen’s data, intellectual property and other proprietary rights and uncertainties relating to intellectual property claims and challenges; product liability claims; third party collaboration risks; and the direct and indirect impacts of the ongoing COVID-19 pandemic on Biogen’s business, results of operations and financial condition. The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from Biogen’s expectations in any forward-looking statement. Investors should consider this cautionary statement as well as the risk factors identified in Biogen’s most recent annual or quarterly report and in other reports Biogen has filed with the U.S. Securities and Exchange Commission. These statements are based on Biogen’s current beliefs and expectations and speak only as of the date of this news release. Biogen does not undertake any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise.
当社のニュースリリースは、企業情報の開示を目的としており、医療用医薬品や開発品のプロモーションや広告、医学的なアドバイスを目的とするものではありません。なお、ニュースリリースに記載している情報は発表日現在のものです。